 |
Tuberous sclerosis complex (TSC) is an inherited, multisystem disorder characterized by hamartomatous growths that can occur in virtually any organ or tissue. Ophthalmic features associated with TSC can be divided into retinal and non-retinal.1 This case highlights a classic presentation of posterior segment findings in a young patient with TSC.
The Case
A 15-year-old male was referred to the eye clinic with no ocular or visual complaints. He had recently been diagnosed with TSC by his primary care provider (PCP) and neurologist, and his PCP wanted to rule out any ocular complications. His history was negative for epilepsy and intellectual disability. Unaided visual acuity was 20/20 OD and OS. Gross exam showed skin adenoma sebaceum along his cheeks extending to his chin. Fundus examination revealed a white, mulberry-shaped lesion in the superior retina of the right eye consistent with retinal astrocytic hamartoma (Figure 1).
We informed the patient’s primary physician about the clinical findings and scheduled the patient for a six-month follow-up visit.
A Bushel of Associations
The term hamartoma refers to abnormal growth of mature cells native to that area of the skin, blood vessels or nervous system. Phakomatoses are a group of hereditary conditions characterized by the presence of hamartomas. Some of the more common phakomatoses, several of which have prominent retinal manifestations, include neurofibromatosis types I and II, tuberous sclerosis (Bourneville’s disease), encephalotrigeminal angiomatosis (Sturge-Weber syndrome), angiomatosis of the retina and cerebellum (von Hippel-Lindau disease) and racemose angioma of the midbrain and retina (Wyburn-Mason syndrome).1,2
The first description of TSC is usually attributed to Bourneville in 1880; however, Vogt described the classic triad of “epilepsy, mental retardation, and adenoma sebaceum” (now called angiofibromatosis) in 1908.2,3
The clinical manifestations of TSC are now known to be more numerous and diverse. TSC is inherited as an autosomal dominant trait—the genes are located on chromosomes 9q and 11q—with an incidence of one in 15,000 live births. There is no race or sex predilection.2,4,5 Systemic manifestations of TSC may include ash-leaf spots, café-au-lait spots, shagreen patches, subungual fibromas, cortical tubers of the basal ganglia, lateral ventricles and third ventricle, obstructive hydrocephalus and tumors of the bone, lung, heart, liver and kidney (Table 1). 5
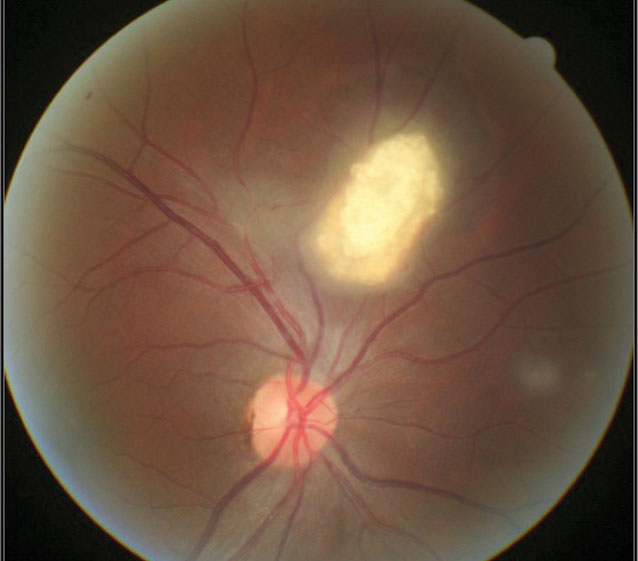 |
| Fig. 1. A mulberry-like lesion consistent with retinal astrocytic hamartoma. |
Ocular Fruits of TSC
Retinal associations of TSC were first noted by Van der Hoeve in 1921.6 He termed these lesions phakomas (derived from the Greek phakos, meaning spot) and introduced the concept of phakomatosis. These lesions are now known to be retinal astrocytic hamartomas (RAH), sometimes called retinal astrocytoma.1,2,4 RAH is a benign, minimally progressive, neoplastic lesion of the retinal nerve fiber layer caused by proliferation of glial cells.
These (typically) elevated yellowish-white calcific “mulberry-like” lesions are the characteristic ophthalmic finding in TSC. RAH is found in 50% to 90% of TSC patients, where they most commonly reside in the posterior pole.1
Three morphological types of retinal hamartomas are described in the literature: (1) the relatively flat, smooth, non-calcified, gray, translucent lesion; (2) the elevated, multinodular, calcified, opaque lesion resembling mulberries; and (3) a transitional lesion that has morphological features of both.2,4,5
Other posterior segment findings associated with TSC include retinal pigmentary disturbances such as hyperpigmented areas (probably congenital retinal pigment epithelium hypertrophy) to “punched out,” hypopigmented areas at the posterior pole or midperiphery.1,2
Other ophthalmic findings include eyelid angiofibromas, iris, lens and choroid coloboma, strabismus, poliosis of eyelashes, optic disc edema and sector iris depigmentation.1,2
Many Hands Make Light Work
Any care provider can make a presumptive diagnosis of TSC if a patient has two of the following: infantile spasms, seizures, areas of increased attenuation in the cerebral cortex, calcified lesion in the cerebral cortex, ash-leaf spots, retinal hamartoma, dental enamel pits and multiple renal tumors.3,5 Clinicians should consult with a pediatric neurologist for a definitive diagnosis.
Patients experience a normal life expectancy. The seizures usually present in infancy and progress to tonic-clonic seizures later in life. A patient with TSC should have an extensive evaluation that includes dermatology, optometry or ophthalmology, neurology, cardiology, urology and pulmonology.5 These subspecialties help ensure a proper interdisciplinary approach to the patient’s care. Some patients may need a shunt procedure for obstructive hydrocephalus.
There is no cure for TSC, although treatment is available for a number of the symptoms. Antiepileptic drugs may be used to control seizures. Sabril (vigabatrin, Lundbeck) is a particularly useful medication and has been approved by the Food and Drug Administration for treatment of infantile spams in TSC, although it has significant side effects.5
Table 1. TSC Review of Systems1-5 |
| Ophthalmic: At least 50% have ocular abnormalities; most common lesions are retinal astrocytomas that become calcified over time. |
| Pulmonary: cystic pulmonary abnormalities occur in up to 40% of women with TSC. |
| Renal: Four types of lesions can occur: autosomal dominant polycystic kidney disease, isolated renal cyst(s), angiomyolipomas (AMLs) and renal cell carcinomas. |
| Dental: Pitting of the dental enamel; gingival fibromas. |
| Gastrointestinal: Hamartomas and polyposis of the stomach, intestine and colon. |
| Hepatic: Hepatic cysts and hepatic AMLs, typically asymptomatic and nonprogressive, with a marked 5:1 female predominance. |
| Skeletal: Sclerotic and hypertrophic lesions of bone. |
| Neurologic: Growth of tubers and the presence of subependymal nodules (SENs) and subependymal giant cell astrocytomas (SEGAs). |
| Cutaneous: Adenoma sebaceum, which often does not appear until late childhood or early adolescence. |
| Cardiac: Of individuals with TSC, 50% to 60% have evidence of early cardiac disease, mostly rhabdomyomas. |
Patients may seek routine eye care with or without an established diagnosis of TSC, and in some cases RAH, detected by the OD, may be the presenting sign of TSC. RAHs usually remain stable over time and have no effect on visual function in the majority of cases. Patients with TSC may experience visual loss secondary to cortical tubers (brain hamartomas), astrocytic hamartomas that involve the macula, optic nerve gliomas and optic atrophy secondary to hydrocephalus.1,2
In patients with a known diagnosis of TSC, clinicians should perform a complete ophthalmic evaluation, including dilated funduscopy, to assess for retinal lesions and visual field deficits. Ongoing periodic surveillance is needed after initial diagnosis for optimal care and prevention of secondary complications. Management of specific complications of TSC will often require input from the multidisciplinary team.2,4,5
The FDA recommends a baseline ophthalmic evaluation and follow-up testing every three months for patients on Sabril.5,7,8 Its use is complicated by an irreversible, dose-dependent visual field constriction from photoreceptor toxicity. Optic neuropathy and macular pigment epithelial changes are the most common ophthalmoscopic findings.7,8 Screening for these in young children can be challenging, especially considering many children who need the medication are non-verbal and often uncooperative. Methods of screening include serial fundus examination, serial automated static perimetry, OCT, VEP and ERG.7,8
Currently in the United States and many other countries, specialized TSC clinics have been established. Ideally, all TSC patients would have access to one of these clinics to ensure appropriate care and treatment. You can help patients locate a clinic at www.tsalliance.org/individuals-families/tsc-clinics.
1. Liu G, Volpe N, Galetta S. Neuro-ophthalmology Diagnosis and Management. Chapter 4 Vision Loss: retinal disorders of neuro-ophthalmic interest. Philadelphia: WB Saunders; 2001:58-102. 2. Rowley SA, O’Callaghan FJ, Osborne JP. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85(4):420-3. 3. Kumar V, Abbas A, Fausto N, Aster J. Robbins and Cotran’s Pathologic Basis of Disease. 7th ed. Chapter 28: the central nervous system. Philadelphia: Elsevier Saunders; 2005: 1347-1420. 4. Quillen DA, Blodi B. Clinical Retina. Chapter 8: intraocular tumors. The American Medical Association. 2002:203-30. 5. Krueger DA, Northrup H. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatric Neurol. 2013;49(4):255-65. 6. Van der Hoeve J. Augengeschwultse bei der tuberosen Hirnsklerose (Bourneville). Albrecht von Graefes Arch Klin Ophthalmol. 1921;105:880-98. 7. Frisen L, Malmgren K. Characterization of vigabatrin-associated optic atrophy. Acta Ophthalmol Scand. 2003;81:466-73. 8. Wild JM, Robson CR, Jones AL, et al. Detecting vigabatrin toxicity by imaging of the retinal nerve fiber layer. Invest Ophthalmol Vis Sci. 2006;47:917-24. |
