 |
During flu season, everyone does their best to steer clear of symptomatic patients. But every once in a while, they need our help. Acute posterior multifocal placoid pigment epitheliopathy (APMPPE), for example, is an immune-mediated chorioretinal disease that usually affects young adults—about 40% of whom report influenza-like symptoms prior to the onset of visual symptoms.1 While uncommon, this white-dot syndrome may end up in your chair. Here’s what you need to know.
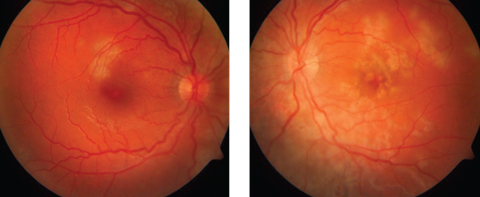 |
| Fig. 1. A-27-year-old female presented one week after a flu-like illness with floaters and blurred vision. Multifocal cream-colored flat placoid lesions were found in the posterior pole of each eye. Click images to enlarge. |
Clinical Features and Course
APMPPE usually involves both eyes, and patients typically present with blur, metamorphopsia or scotomas with characteristic fundus findings. The mean age of onset is 27 years and it affects males and females in equal numbers.2
Although the exact cause of APMPPE is unknown, researchers suspect a virus may be to blame. For one, the condition can subside without treatment and may recur at any time, similar to other conditions with a viral etiology. Viruses may stay dormant for extended periods of time, then for reasons yet unknown may unexplainably become reactivated.1,5
In addition, prior to the onset of APMPPE, patients may present with virus-like symptoms such as nausea, fever, swollen lymph glands and vomiting. Moderate to severe headaches may also occur and, rarely, patients may have neurological signs such as aphasia (temporary loss of speech), limb weakness or both.1,2 In the early stages of the disease, patients may complain of blotchy scotomata, photopsia, metamorphopsia and photophobia. Later stages are marked by moderate decreases in vision. Less commonly, the impaired vision may be severe.3
APMPPE may present with anterior segment findings that include episcleritis, non-granulomatous uveitis and perilimbal anterior stromal corneal infiltrates. The vitreous may have a mild cellular reaction that usually accompanies multifocal yellowish-white flat placoid lesions located mainly in the posterior pole involving the retinal pigment epithelium (RPE) (Figure 1).4 These lesions do not extend beyond the equator and tend to fade over a two-week period, where they are replaced by varying degrees of RPE atrophy and hyperpigmentation.1,4
Other findings may include papillitis, retinal periphlebitis, central retinal vein occlusion, optic nerve neovascularization and subhyaloid hemorrhage.
Intravenous fluorescein angiography (IVFA) will show a classic “block early, stain late” pattern.1-4 The early phase of IVFA will show the acute lesions as hypofluorescent, suggesting nonperfusion or infarction of the RPE, choroid or both (Figure 2). The lesions then become hyperfluoresent in the late phase of the study (Figures 2 and 3).1,2
Optical coherence tomography findings in APMPPE include subretinal fluid with a hyperreflective line anterior to the RPE.1
Table 1. Differential Diagnosis of APMPPE | |
| Type | Condition |
| Other white-dot syndromes | • Serpiginous choroiditis • Multifocal choroiditis and panuveitis • Punctate inner choroidopathy • Birdshot chorioretinopathy • Multiple evanescent white-dot syndrome |
| Infectious conditions | • Syphilis • Tuberculosis • Fungal disease • Toxoplasma retinochoroiditis • Pneumocystis choroiditis • Viral retinitis |
| Neoplastic disease | • Choroidal metastases • Lymphoma |
Systemic Concerns
A review of systems is important in cases of suspected APMPPE, as systemic associations of APMPPE may involve the skin (erythema nodosum), kidneys (nephritis with urine casts), muscles, thyroid gland (thyroiditis) and blood vessels (vasculitis).
It has also been associated with multiple complications in the central nervous system (CNS), including cerebral vasculitis, peripheral neuropathy, headaches, aseptic meningitis, meningoencephalitis, sixth cranial nerve palsy, transient hearing loss and cavernous sinus thrombosis.1,3 Symptoms of severe headache or meningeal symptoms warrant neuroimaging and further neurologic workup. Research has associated fatalities due to cerebral vasculitis with the disease.1
APMPPE-like lesions can be present in patients with sarcoidosis, syphilis and tuberculosis; therefore, clinicians should test patients to exclude those conditions.4
Differential Diagnosis
The white-dot syndromes are a group of multifocal inflammatory conditions involving both the retina and the choroid. They are characterized by the appearance of white dots in the fundus. Several of these conditions can simulate APMPPE (Table 1).
The most similar of the white-dot syndromes to APMPPE is serpiginous choroiditis. The chorioretinal lesions in serpiginous choroiditis are localized to the posterior pole, but produce a more profound choroidal atrophy than in APMPPE. Serpiginous choroiditis also resolves more slowly than APMPPE, and patients have a poorer visual prognosis, with more frequent recurrences of inflammation.3,4
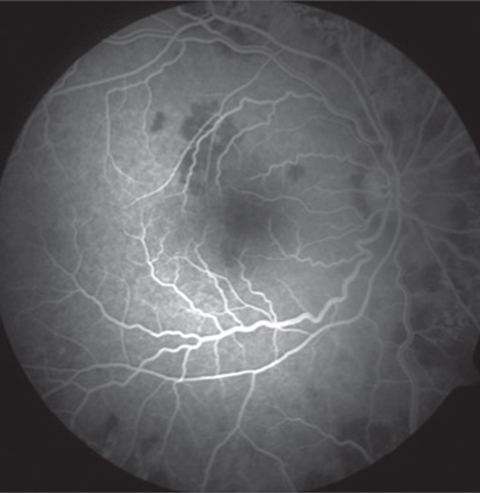 | 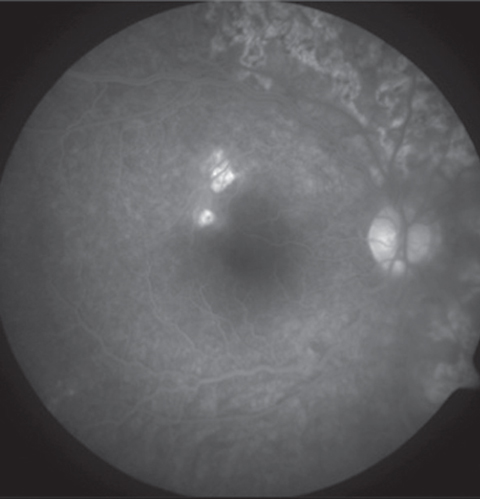 |
| Fig. 2. Early phase of the IVFA shows the acute lesions are hypofluorescent. | Fig. 3. In the late phase of the study, the lesions become hyperfluorescent. |
Management
Most cases of APMPPE resolve within a few weeks. However, in some cases where central macular involvement is profound, visual acuity does not significantly improve.2
Because APMPPE is generally self-limiting, there is no rationale for treatment if no neurological complication is encountered. The outcome for the visual system without treatment is characteristically good, and a gradual improvement in visual acuity occurs over weeks to months, with most eyes achieving a visual acuity of 20/30 or better.
If CNS vasculitis is present, systemic corticosteroid treatment is recommended. Steroid therapy may also be indicated for extensive disease that involves the fovea. This may offer a theoretical advantage in shortening the disease course or modifying its effects on central vision.1,6
APMPPE is an immune-mediated disease characterized by discrete areas of subretinal inflammation. It may be associated with a number of systemic conditions and thus warrants a detailed systemic workup. Although the disease is self-limiting with a relatively good prognosis, patients with extensive macular involvement may be treated with systemic steroids in an effort to preserve visual acuity to the greatest extent possible.
1. Algahtani H, Alkhotani A, Shirah B. Neurological manifestations of acute posterior multifocal placoid pigment epitheliopathy. J Clin Neurol. 2016;12(4):460-7. |
